
MDR transition
Regulation (EU) 2017/745 on medical devices (MDR) has replaced the dated Medical Device Directive 93/42/EEC (MDD) to help increase overall compliance and standards for the quality and safety of medical devices marketed in the EU as well as to raise public awareness and ensure continuous monitoring of product safety and performance throughout the entire product lifetime.
MDR requirements are significantly more extensive than the MDD: while the MDD comprises 23 Articles and 12 annexes over 60 pages, the MDR has 123 articles and 17 annexes over 175 pages. Therefore, to maintain or obtain CE certification, companies must meet MDR requirements to remain in compliance and keep their product on the EU market.
Extension of the transition period
By June 2022, it was clear that compliance with the transitional phase, which was set for May 26th, 2024, was unlikely to occur since the market and industry faced numerous difficulties that would ultimately impact consumers. The biggest challenge was the availability and capacity of notified bodies (NB) accredited under MDR; post-Brexit, the EU no longer recognizes UK Notified bodies, eliminating their right to issue CE certificates, thus diminishing the number of Notified bodies available to help with the transition.
Other contributing factors include lack of advanced preparation and planning by manufacturers due to uncertainties surrounding NB availability, EUDAMED, lack of trained resources, new compliance expectations imposed by MDR, e.g., additional safety and performance data, i.e., General Safety and Performance Requirements (GSPRs), plausible reclassification of several devices into higher risk categories, among others. Hence, taking these concerns into account, on March 15th, 2023, the EU enacted Regulation (EU) 2023/607, which provides a much-needed extension to the original MDR transitional provisions.
The extensions only apply to devices that have not undergone significant change in design or intended use and do not present an unacceptable risk to users and patients, and for which manufacturers have taken the necessary steps to comply. The fact that the MDR transition period has been extended is good news for manufacturers, but there isn’t really much time to relax.
New MDR Compliance Timeline
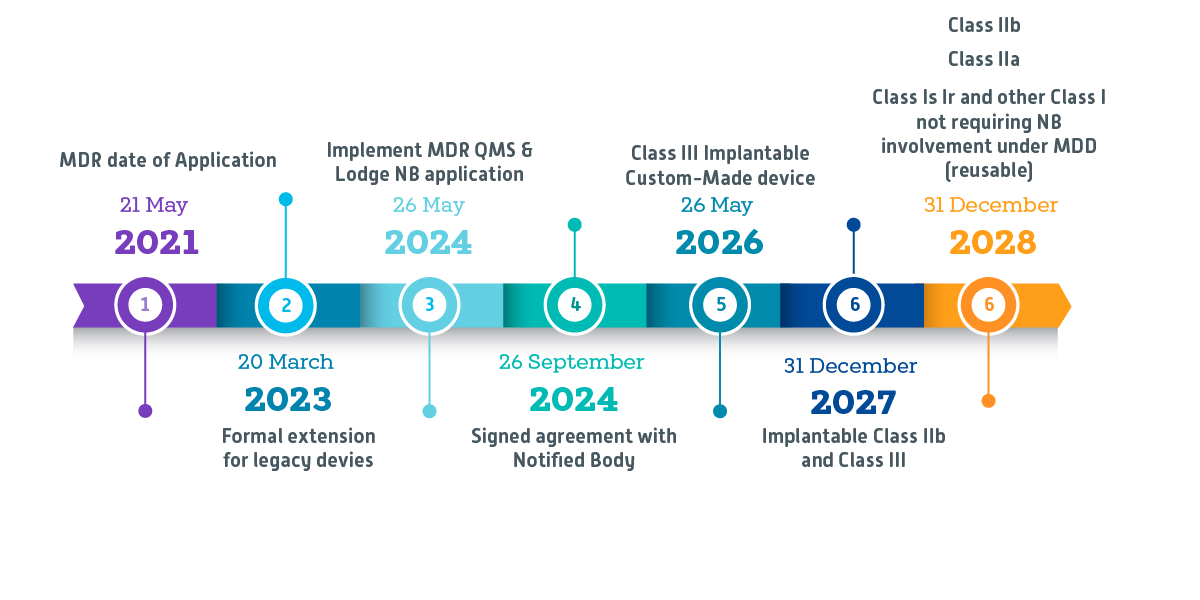
What should Manufacturers do?
Manufacturers should have a valid MDD/AIMDD certificate on the MDR Date of application (May 26th, 2021) and completed the following by May 26th, 2024, to qualify for the extension:
1. Implement a quality management system as outlined in MDR Article 10(9) and
2 . Formally lodge an application with a Notified Body for the device
Note: QMS implementation by this timeline is exempted for Class III implantable custom-made devices
The only exception to this would be if the competent authority has approved a derogation under Article 59 MDR or 54 IVDR. It is therefore imperative for continual business that manufacturers complete their due diligence, rather than assuming that their products will automatically be extended under the MDR if they comply with requirements after adoption. Failure to comply with the above timelines and regulations prevents the device qualification under this extension.
ClinChoice’s support and services
MDR’s scope of change will necessitate effective project management to establish that tasks are properly completed on schedule.
At ClinChoice, we provide services to expedite and simplify compliance achievement at any stage of the transition and compliance to help ensure our clients are not impacted from a business standpoint and that the transition process is seamless.
Our experts use their skills to create a transition plan and see it through to completion by implementing best practices and leading project teams at each stage. Our support includes but is not limited to – meeting the checklist of MDR, classification review, gap assessment, setting up the clinical evaluation process, technical documentation compilation, post-market vigilance system establishment, assisting companies to qualify for extension timelines by lodging a formal application, and written agreement, among others.
About Us
ClinChoice is a leading global Contract Research Organization (CRO), with over 3,700 clinical research professionals across North America, Asia, and Europe. For more than 27 years, ClinChoice has been providing high-quality contract research services to pharmaceutical, biotechnology, medical device, and consumer products clients, encompassing a broad range of services and therapeutic areas. ClinChoice offers cutting-edge, full-service solutions for Clinical Trials, Regulatory Affairs, Medical Device Safety, Toxicology, and Medical Affairs.
References
- EU Commission Q&A Pamphlet
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745
- https://www.gov.uk/guidance/regulating-medical-devices-in-the-uk
- https://health.ec.europa.eu/system/files/2023-07/mdr_proposal_extension-q-n-a.pdf