
Pertaining to the radiodiagnostic agents, there are few changes in the non-clinical requirements between the EMA (European Medical Agency) (EMA/CHMP/SWP/686140/2018) [1] and US FDA (United States Food and Drug Administration) (Docket Number: FDA-2017-D-5297, Document ID: FDA-2017-D-5297-0007) [2]. The requirements are not the same between them; therefore, for someone who needs to register a product through EMA and US FDA, it is necessary to know about the prerequisites.
The objective of this article is to summarize the EMA and US FDA non-clinical requirements for the registration of radiodiagnostic agents, along with comparing and comprehending the similarities and differences between them.
EMA versus US FDA, non-clinical requirements for radiodiagnostics [1,2]
The non-clinical requirements for registration of radiodiagnostic agents in EMA and US FDA are illustrated in Table 1.
The non-clinical phase is one of the important aspects of drug development; hence, extensive testing is required before the first human exposure. Specific to radiodiagnostics, the non-clinical development requirements are relatively lesser as compared to the non-clinical development of a drug; however, the type of non-clinical studies to be conducted should be determined on a case-by-case basis. For instance, the US FDA has discovered delayed radiation effects of radiodiagnostics as a serious risk, and such issues can be addressed through dosimetry and radiation threshold studies in animals before first human exposure. Because of the small dose (microdose), infrequent exposure (unless it is warranted), and low potential for toxicity compared to pharmaceuticals and radiotherapeutics.
In these lines, when an inventor is looking to launch an already existing radiodiagnostic product in a different geographical region (For example, for launching a USFDA-approved radiodiagnostic agent in EMA region or vice versa), based on the regulatory requirements, gap analysis needs to be performed to ascertain further steps on the submissions. Knowing the regulatory requirements (EMA and USFDA) will help us develop the checklist for the dossier preparation.
At Clinchoice, we have a dedicated team of experts who look after the different aspects of radio diagnostic submissions for approval, which includes summarizing the regulatory requirements, performing gap analysis, developing checklists and regulatory strategies, preparing of dossier, among other services.
Table 1. Illustration of Non-clinical development requirements for US FDA and EMA
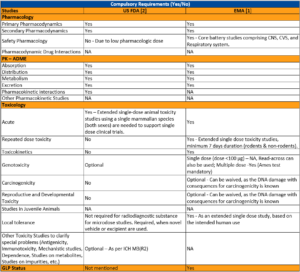
NA – Not applicable; CNS, Central nervous system; CVS, Cardiovascular system.
Talk with a specialist today to learn how ClinChoice can support and alleviate hurdles in your submission.
About Us
ClinChoice is a leading global Contract Research Organization (CRO), with over 3400 clinical research professionals across North America, Asia, and Europe. For more than 27 years, ClinChoice has been providing high-quality contract research services to pharmaceutical, biotechnology, medical device, and consumer products clients, encompassing a broad range of services and therapeutic areas. ClinChoice offers cutting-edge, full-service solutions for Clinical Trials, Regulatory Affairs, Medical Device Safety, Toxicology, and Medical Affairs.
Authors
Dr. GL Vishwanatha, M. Pharm, Ph.D., ERT (UK-RT), PG (Statistics), MBA
Associate Manager – Toxicology team
Clinchoice Private Limited
Bangalore-560078
Karnataka, India
E-mail: vishwanatha.gl@clinchoice.com
Phone: 098444 92334
Mr. Ashish Kapoor, M. Sc, DABT, ERT (UK-RT)
Associate Manager – Toxicology team
Clinchoice Private Limited
Bangalore-560078
Karnataka, India
E-mail: ashish.kapoor@clinchoice.com
References: